Tutorial
1 Euphorbiaceae DB home page

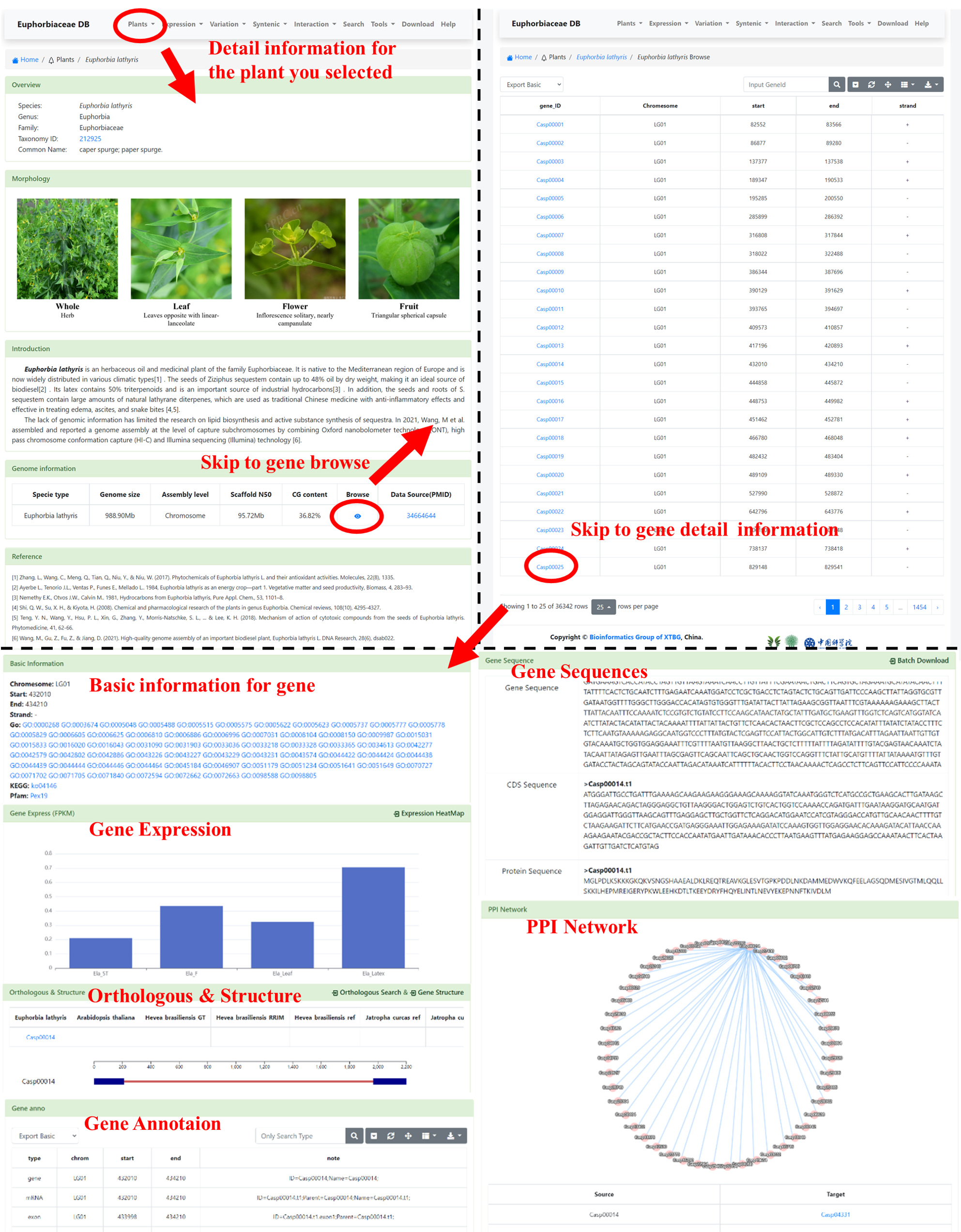
2 Plant
3 Expression
3.1 Expression Heatmap
You can use this tool to obtain the expression levels of protein-coding genes in 12 genome sets of 6 species of Euphorbiaceae under different tissue or stress conditions. User needs to select the interested genome, inputs the Gene ID (multiple inputs are supported, separated by commas), choose Heatmap Normalize (4 options) and select the tissue or stress condition (multiple selections are supported). After submission, the expression levels (FPKM) of the input genes were retrieved in a tabular format and visualized as a heatmap.

3.2 miRNA Target Prediction
We downloaded 708 miRNA sequences of 4 species (Ricinus communis, Jatropha curcas, Manihot esculenta, and Hevea brasiliensis) from the PmiREN, and then used the online tool psRNATarget to predict the miRNA target sites of these genomes. User needs to select the interested genome, inputs Gene ID (if don't input anything we will show all in the genome by default), and finally selects the miRNA sequence. After submission, we will show a form, including Gene ID, miRNA ID, Transcript ID, Expected Value, miRNA Seq and Target Seq. This form supports retrieval by Gene ID and provides functions such as downloading. Click on the Gene ID to jump to the related detail page.

3.3 DNA Methylation
We provide a visual view of DNA Methylation for 3 accession in Ricinus communis(ZB306) and Manihot esculenta(AMS560-2, TME-7), and click to Jbrowse page.
,

4 Variation
4.1 SNP
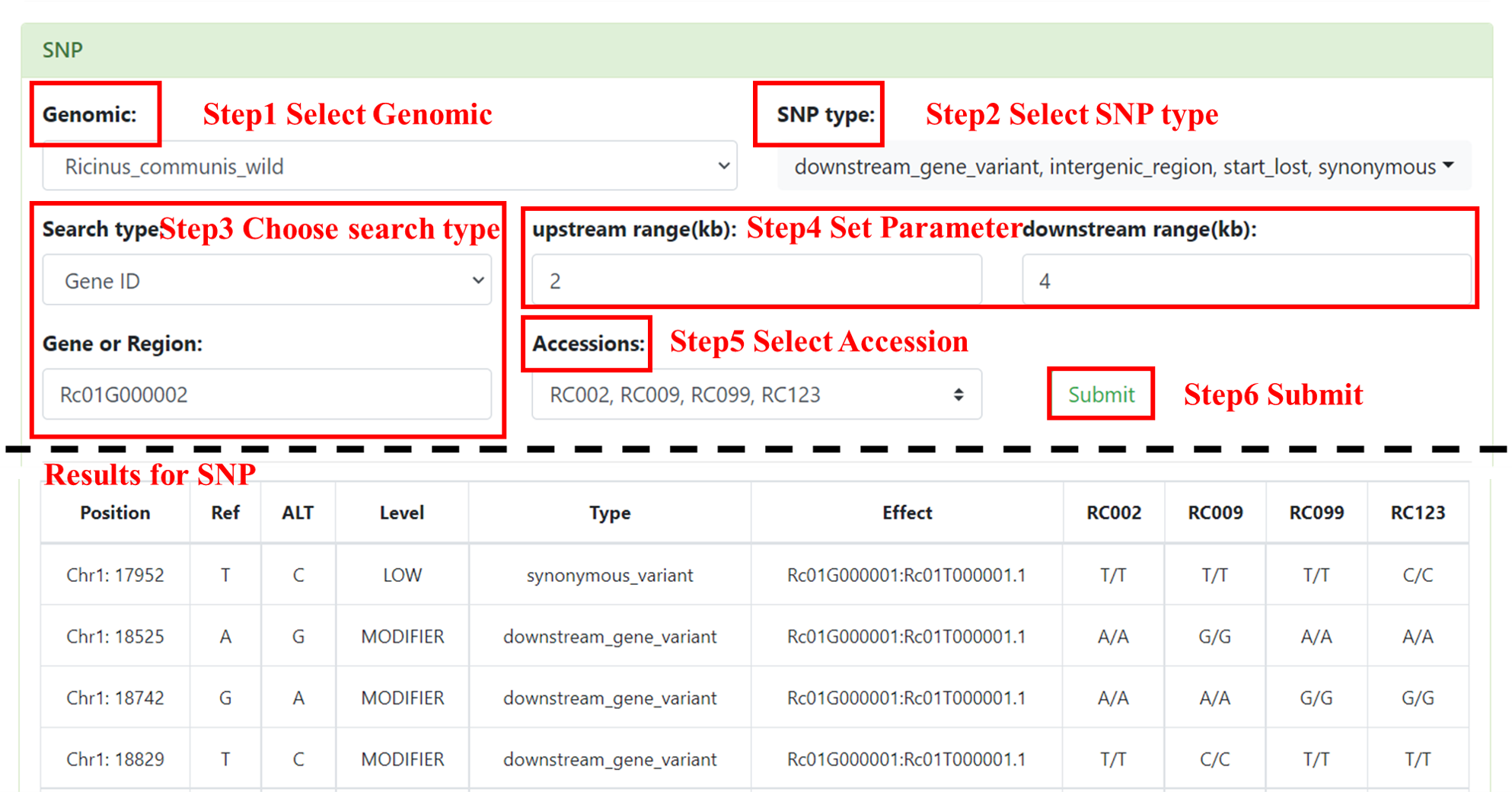
You can obtain the SNP information of the interested accessions. User needs to select the interested Genomic and the SNP type, chooses the Search type (Gene ID and Region), sets the range (default is 0, and the maximum value does not exceed 50kb), enters Gene or Region and selects Accessions. After submission, the table shows SNP information of the input, including SNP Position, Ref, Alt, Level, Type, Effect and the variant information of each accession.
4.2 GWAS & QTL
You can obtain the interested agricultural traits of GWAS or QTL. User needs to select the interested Genomic, Search type and Trait. After submission, the table shows related genomic position and support a quick link to SNP page.

4.3 LDheatmap
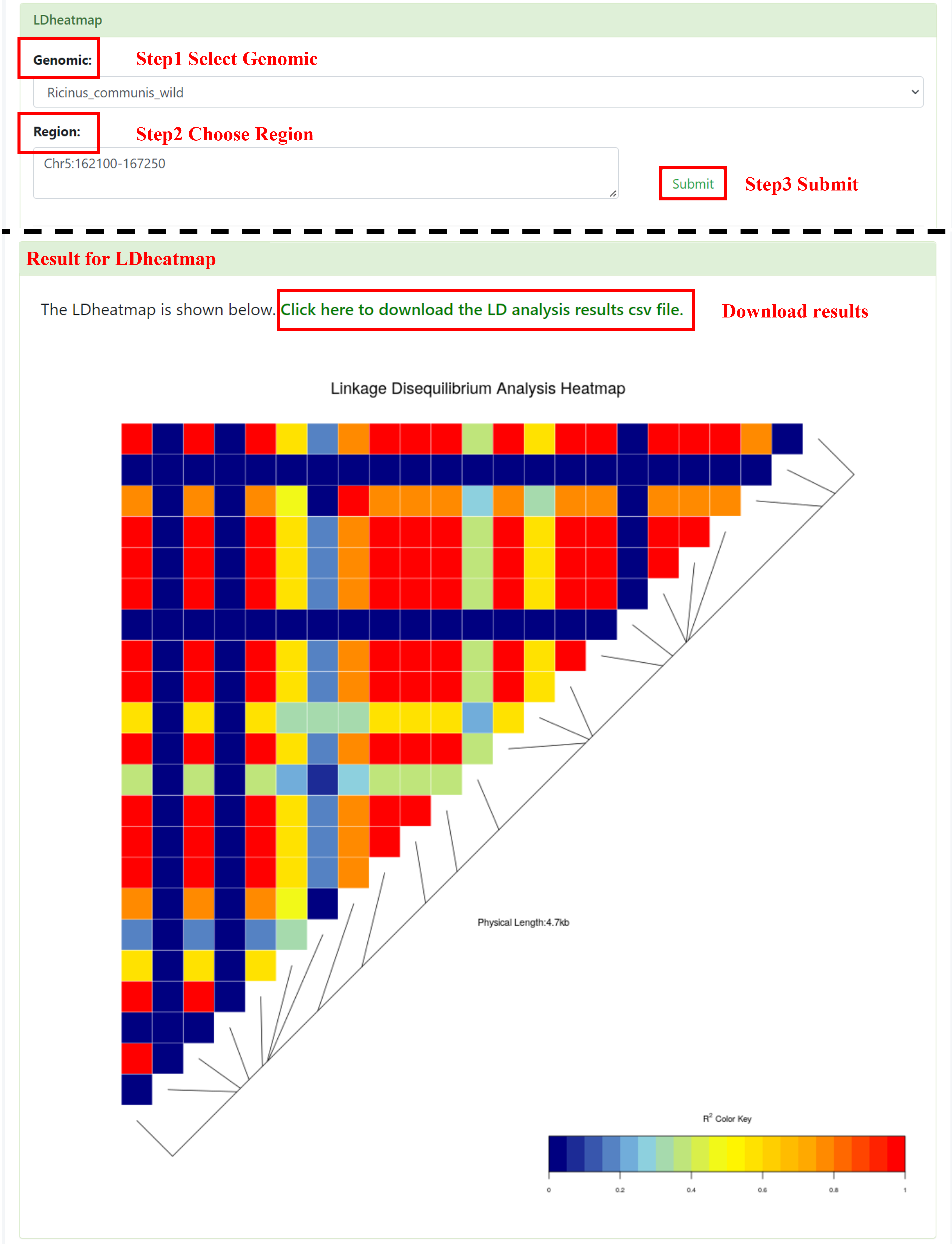
User needs to select the interested Genomic and enter Region. After submission, we will show a picture visualized as a linkage disequilibrium heatmap.
5 Syntenic
5.1 Syntenic Gene
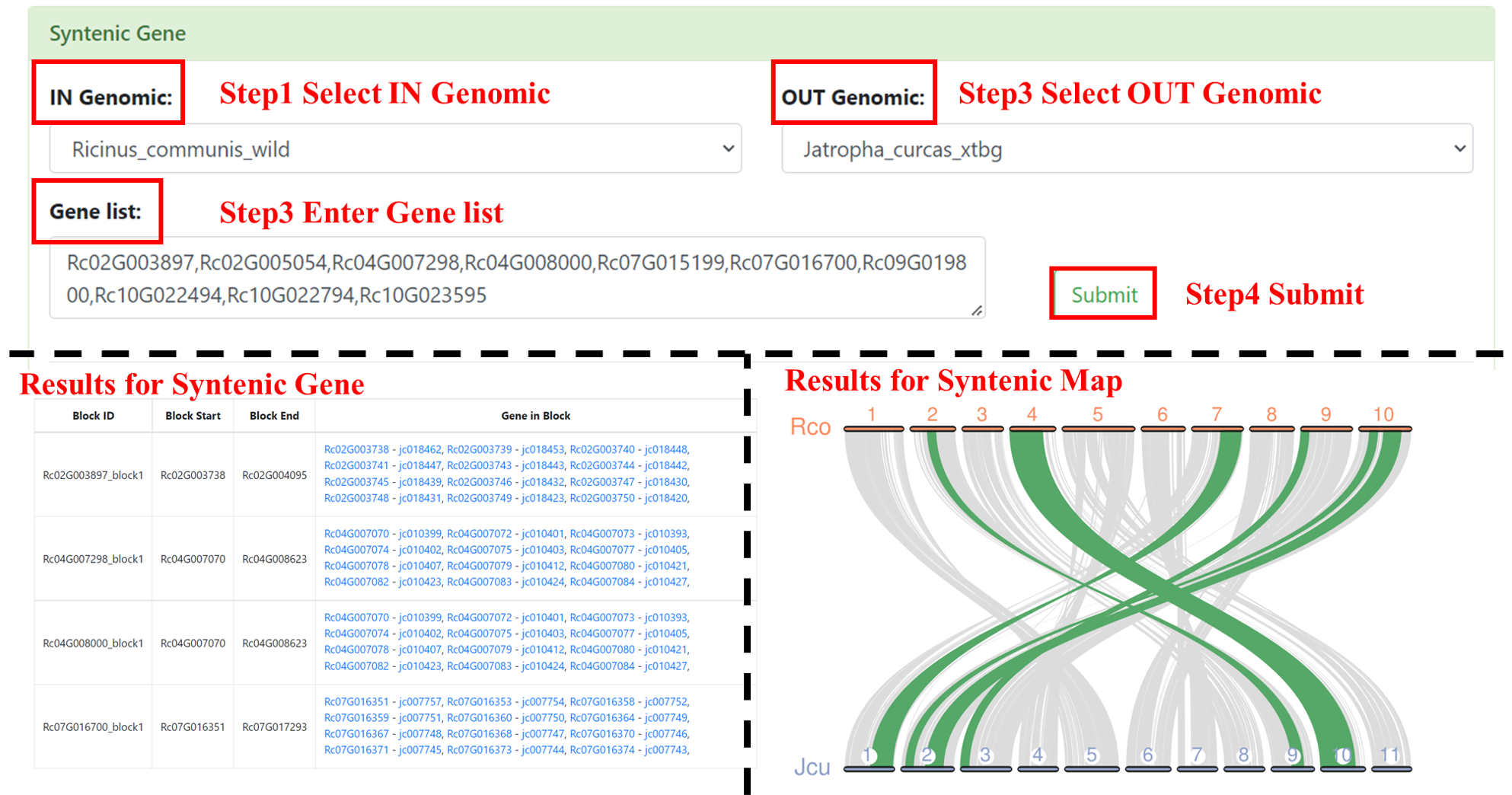
You can use Syntenic Gene to find homologous genes. Users need to select the genome in IN Genomic to find the genes of collinear fragments, then select the genome to compare in OUT Genomic, and finally enter the genes of the species to be compared. We will return the table and collinearity plot to you.
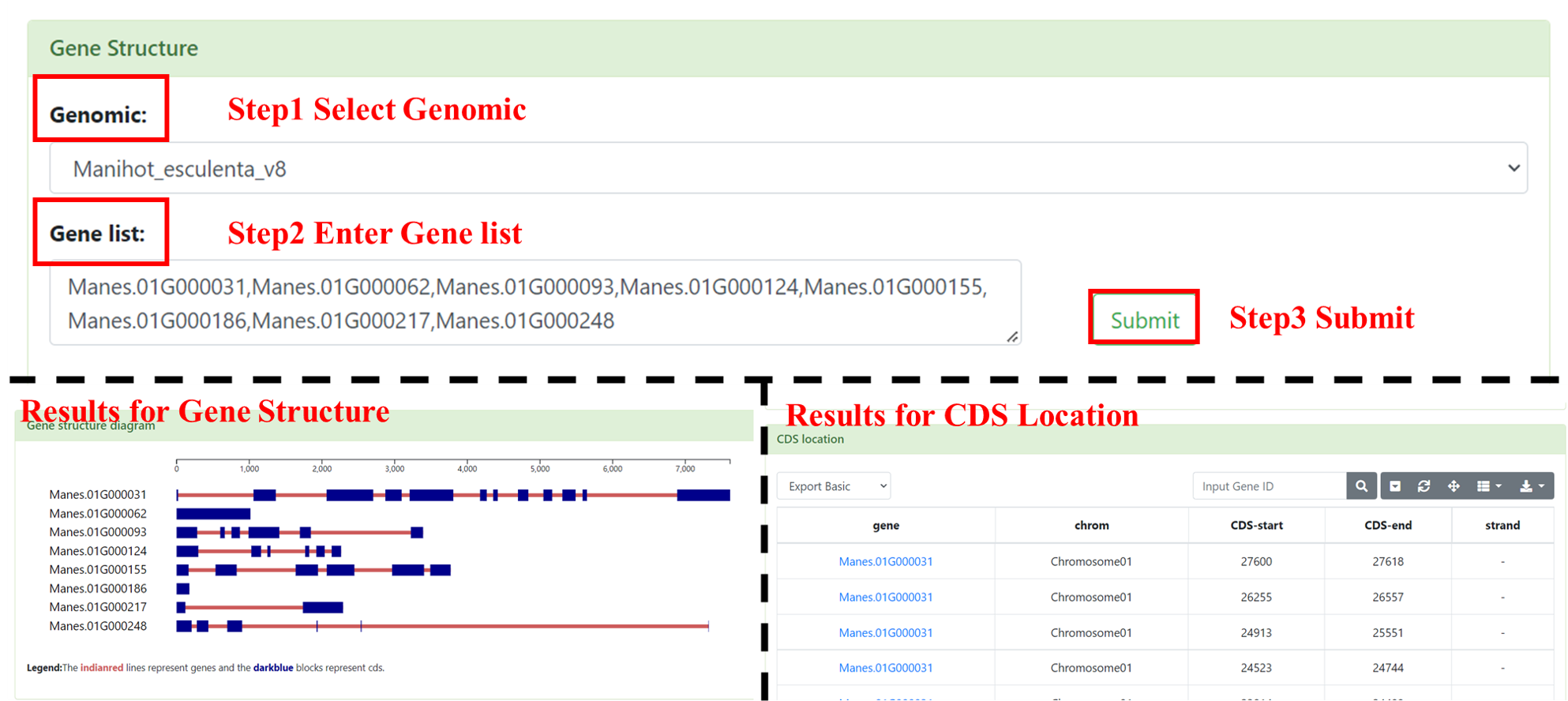
5.2 Gene Structure
You can use Gene Structure to obtain the interested linear structure and CDS location of genes. User needs to select the interested genome and inputs the Gene ID (multiple inputs are supported, separated by commas). After submission, we will show CDS locations information as a form and visualized the gene linear structure map.
5.3 Orthologous Groups
We used BLASTP to obtain the best alignments between species, providing a search for homologous sequences between the user querying species. After submission, we will show a form, including the best genes obtained by comparing the output genome with the input genome in OUT Genomic. This form supports retrieval by Gene ID and provides functions such as downloading. Click any Gene ID to jump to the related detail page.

6 Interaction
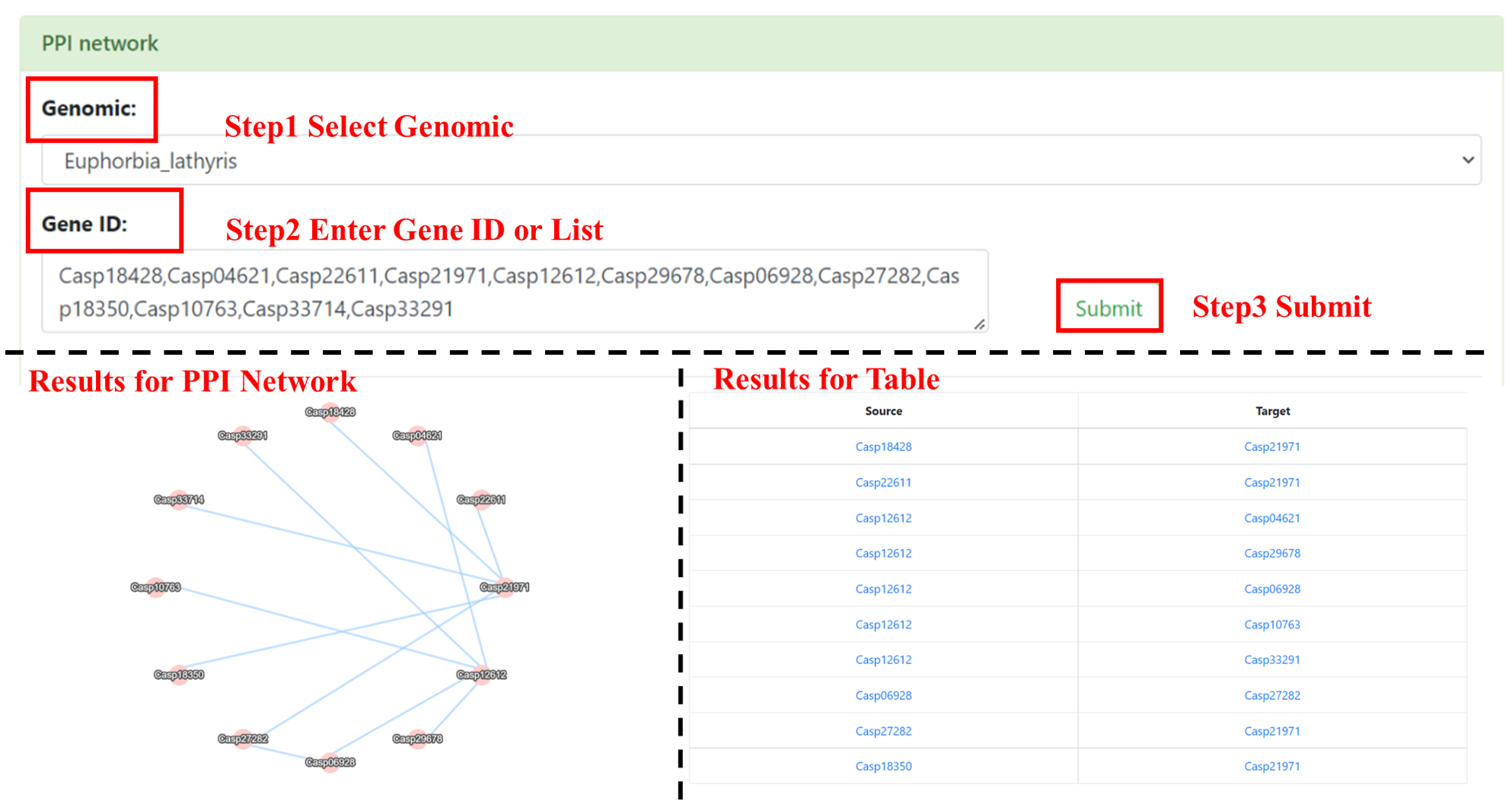
6.1 PPI Network
Users need to select interested genomes and enter Gene ID or list. After submission, the results is a network which shows the interaction between input genes.
6.2 Pathway
You can use Pathway to predict functional genes in 19 hot pathways in Euphorbiaceae species. Users need to select pathways and interested genomes (multiple selections are supported). The prediction results are based on the Arabidopsis genes that function in the selected pathway as a reference (green in the pathway map represents the functional Arabidopsis genes), and the list shows the optimal genes predicted to function in the pathway (the best results of two-way comparison). If users need to get more prediction results, you can use the BLAST to compare.

7 Search
7.1 Keywords Search
The Keyword Search provides 4 search functions, including Gene Search, Go Search, KEGG Search, and Pfam Search. The user needs to select the interested genome and Search type, then enter the keywords ID or list. After submission, we will show a form that supports retrieval by gene ID and provides functions such as downloading. Click on the Gene ID to jump to the gene detail page.

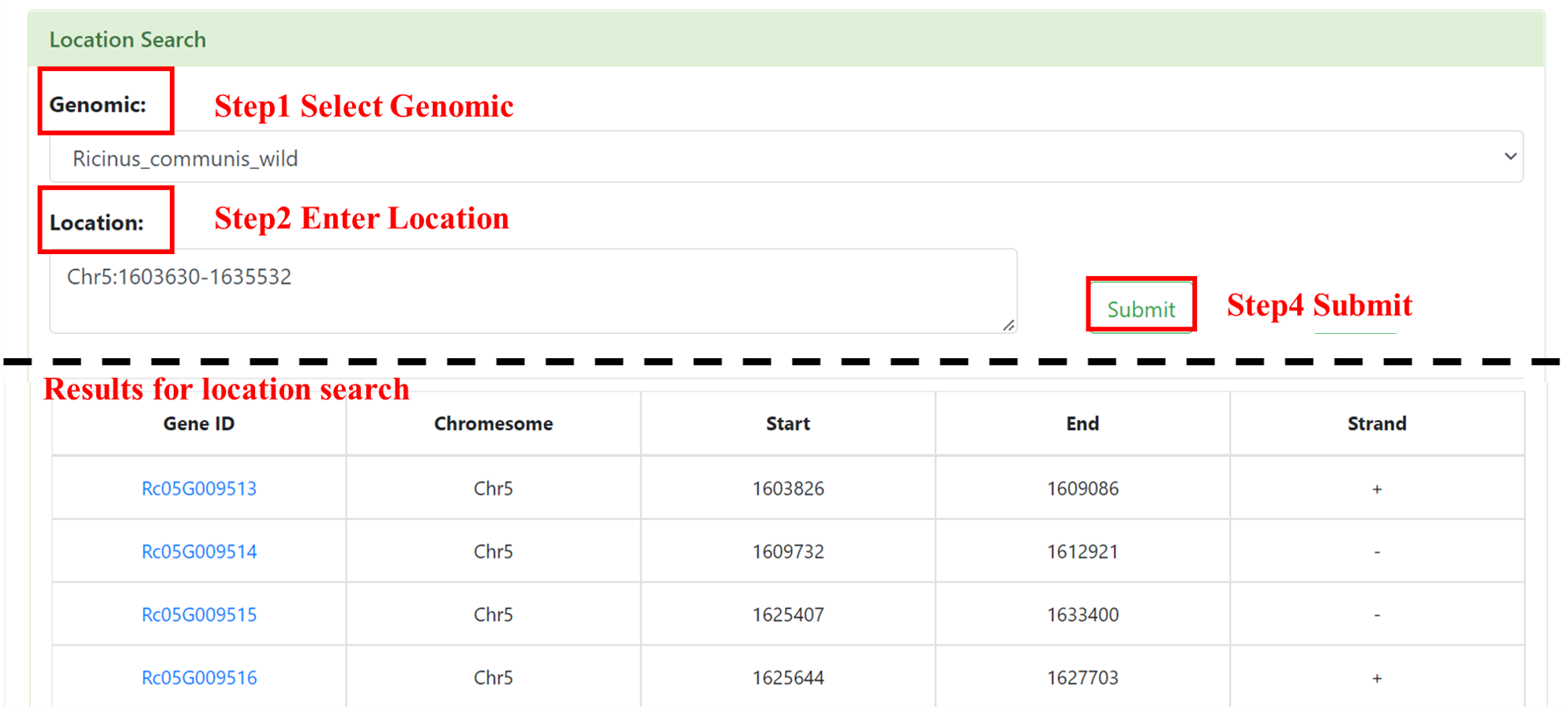
7.2 Location Search
Users can select the interested genome and enter the location (chromosome: start position-end position). After submission, we will show a form, including Gene ID, Chromesome, Start, End and Strand. This form supports retrieval by gene ID and provides functions such as downloading. Click on the Gene ID to jump to the gene detail page.
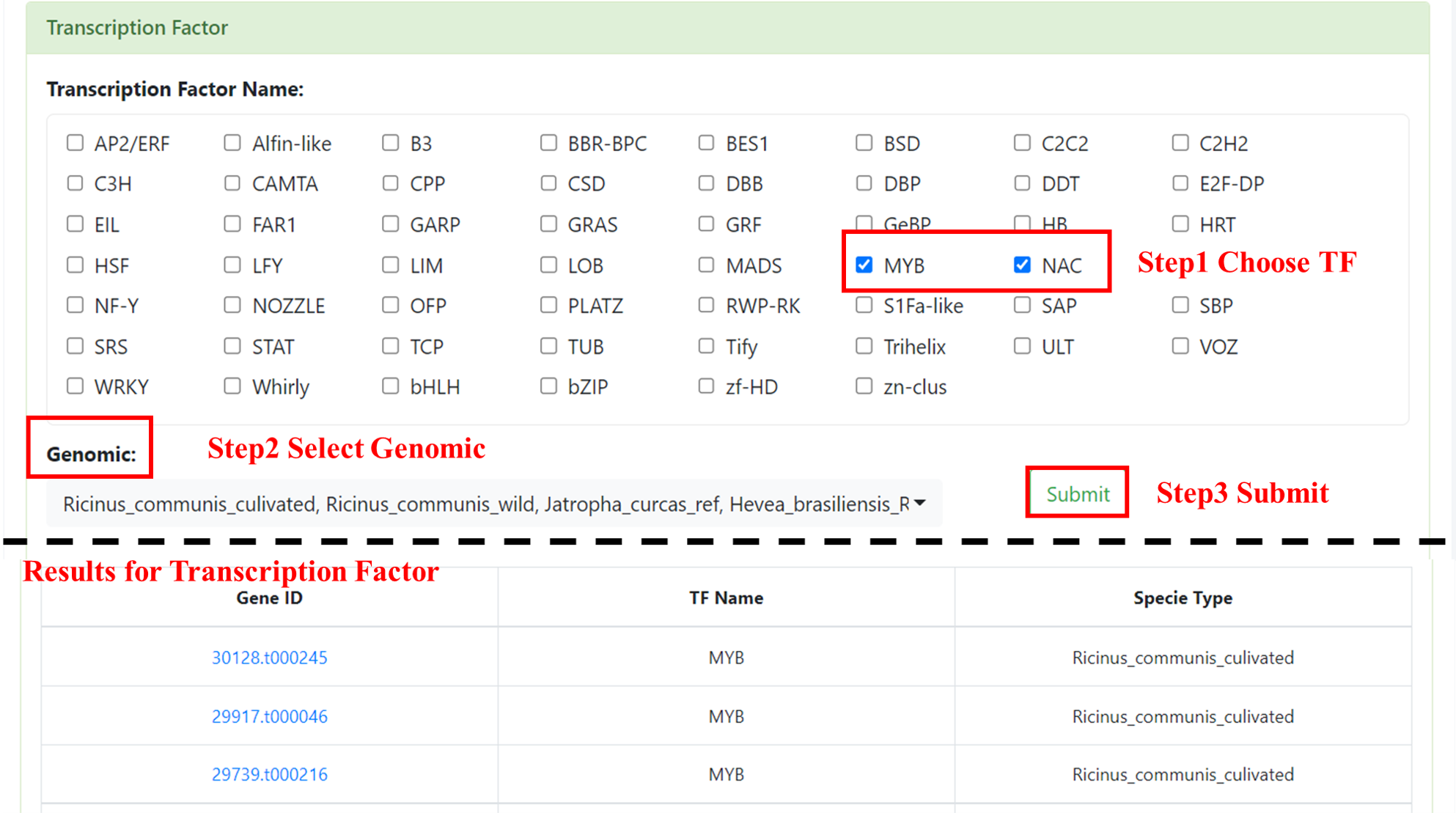
7.3 Transcription Factor
We provide 54 Transcription Factors, users can select one or more TFs, and the interested genome (multiple selections are supported). After submission, we will show a form, including Gene ID, TF Name and Specie. This form supports retrieval by gene ID and provides functions such as downloading. Click on the Gene ID to jump to the gene detail page.
7.4 Transcription Regulator
We provide 24 Transcription Regulators, users can select one or more TRs, and the interested genome (multiple selections are supported). After submission, we will show a form, including Gene ID, TR Name and Specie. This form supports retrieval by gene ID and provides functions such as downloading. Click on the Gene ID to jump to the gene detail page.

8 Tools
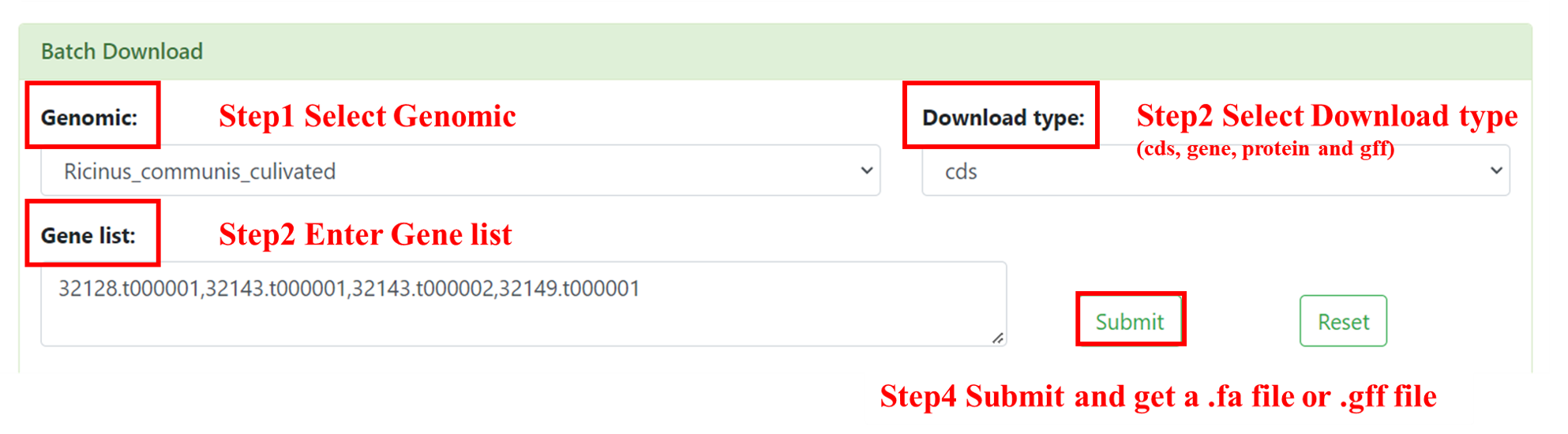
8.1 Batch Download
You can use this Batch Download to batch download you interested CDS, gene or protein sequences. User needs to select the interested genome, inputs the Gene ID (multiple inputs are supported, separated by commas) and selects download type (cds, gene and protein). After submission, you will get a .tsv file.
8.2 BLAST
You can use BLAST to find homologous sequences in EupDB of interested gene. User needs to enter the fasta format sequence in the search box or directly drags the file into the search box, selects the Blast database to be compared, and sets the search parameters (optional, the default is evalue: 1.0e-5; num_alignments: 100 ), select software to use (BLASTN, BLASTP, TBLASTX and TBLASTN are supported). After submission, we will shows the results of the BLAST alignment.

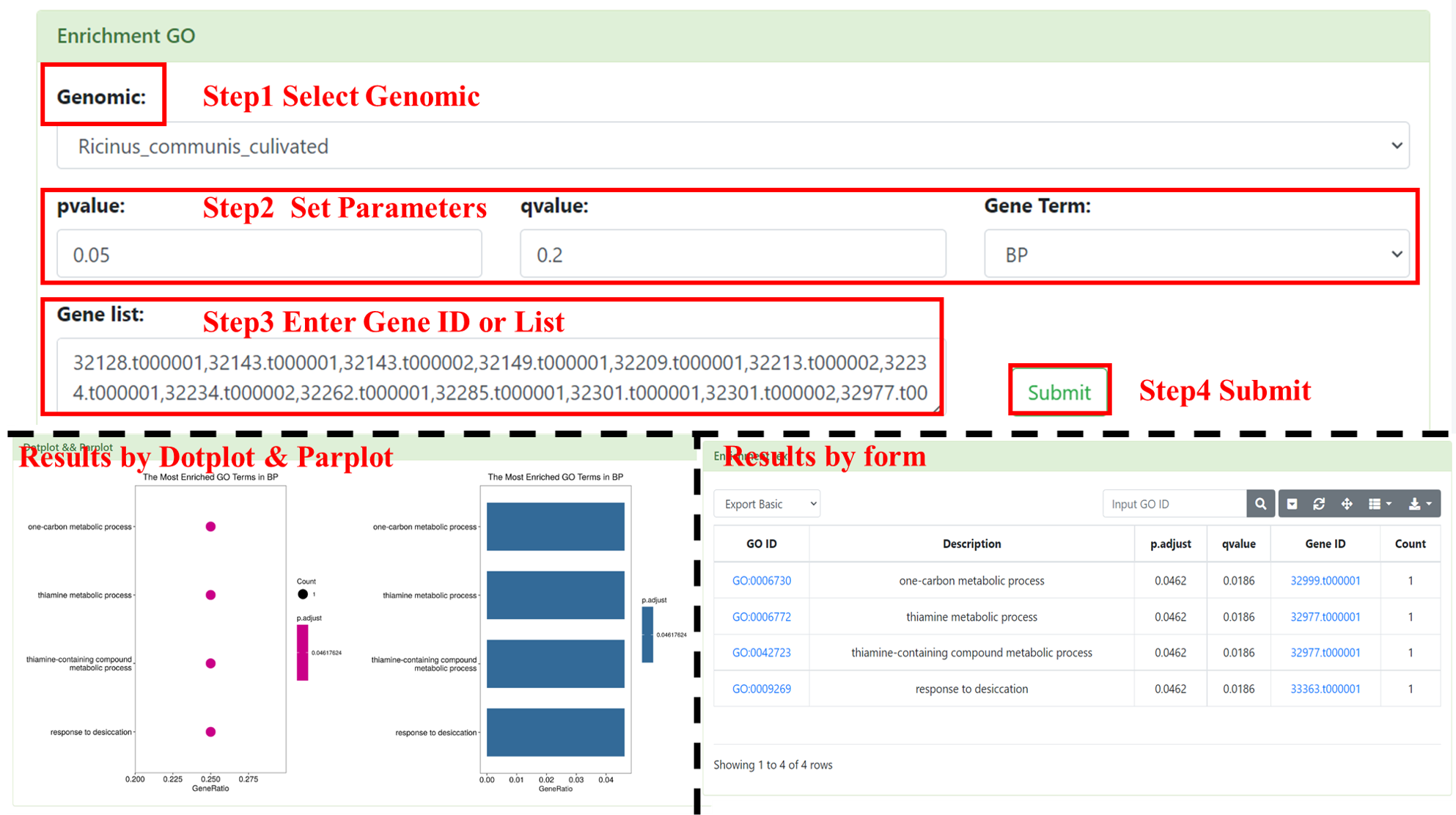
8.3 Enrichment GO
You can perform GO enrichment analysis online. User needs to select the interested genome (multiple selection are supported), inputs Pfam ID (multiple inputs are supported, separated by commas), set the pvalue and qvalue parameter and GO Term (BP, CC, and MF). After submission, we will show a table and figures (Dotplot and Parplot). The table includes GO ID, Description, p.adjust, qvalue, Gene ID and Count. This table supports retrieval by GO ID and provides functions such as downloading.
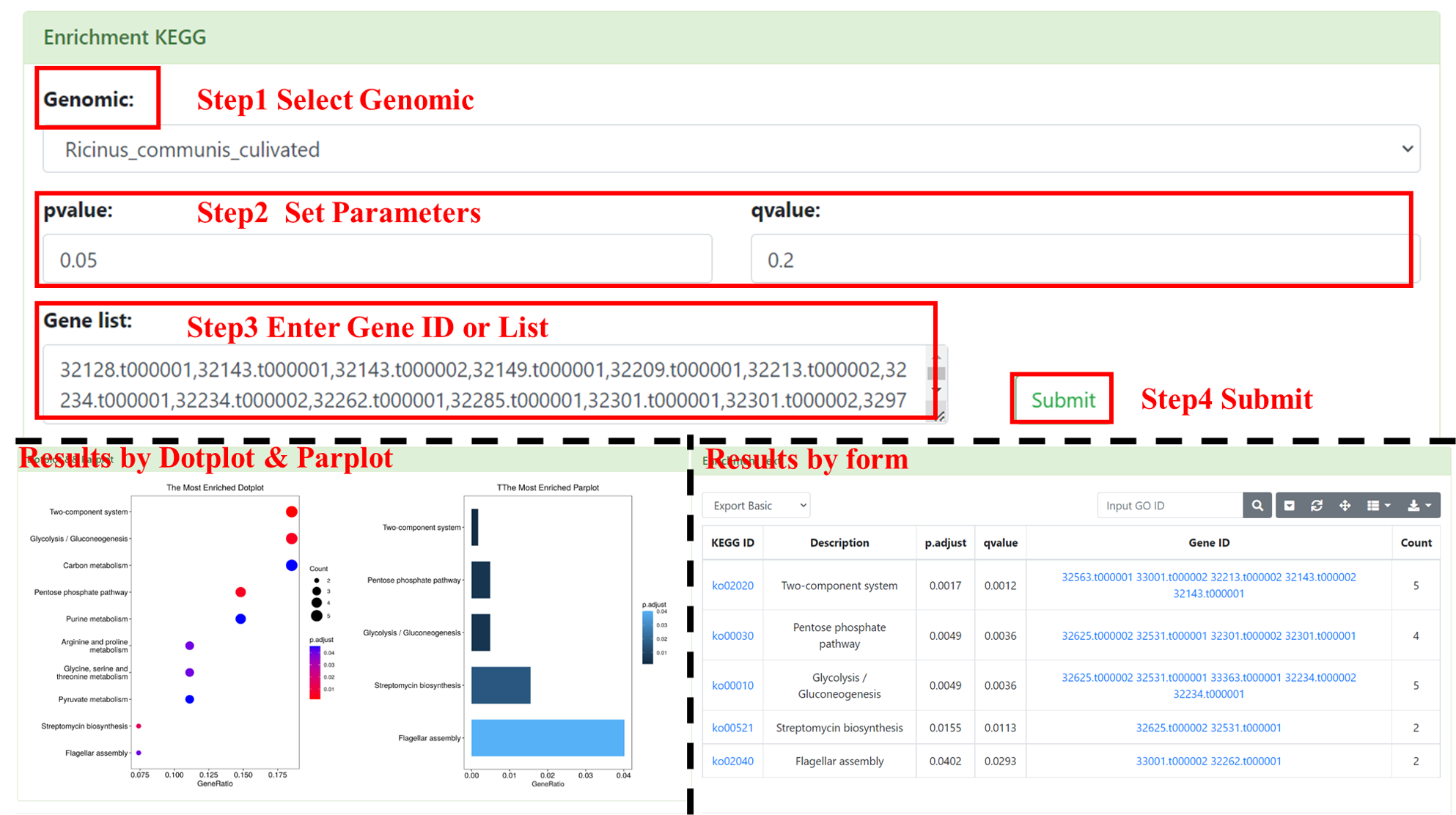
8.4 Enrichment KEGG
You can perform KEGG enrichment analysis online. User needs to select the interested genome (multiple selection are supported), inputs Pfam ID (multiple inputs are supported, separated by commas) and set the pvalue and qvalue parameter. After submission, we will show a table and figures (Dotplot and Parplot). The table includes KEGG ID, Description, p.adjust, qvalue, Gene ID and Count. This table supports retrieval by KEGG ID and provides functions such as downloading.
8.5 Sequence Get
You can use Seq Get to get sequences that you interested. User needs to select the interested genome and input interested gene location. After submission, you will get a .fa file.

9 Download
